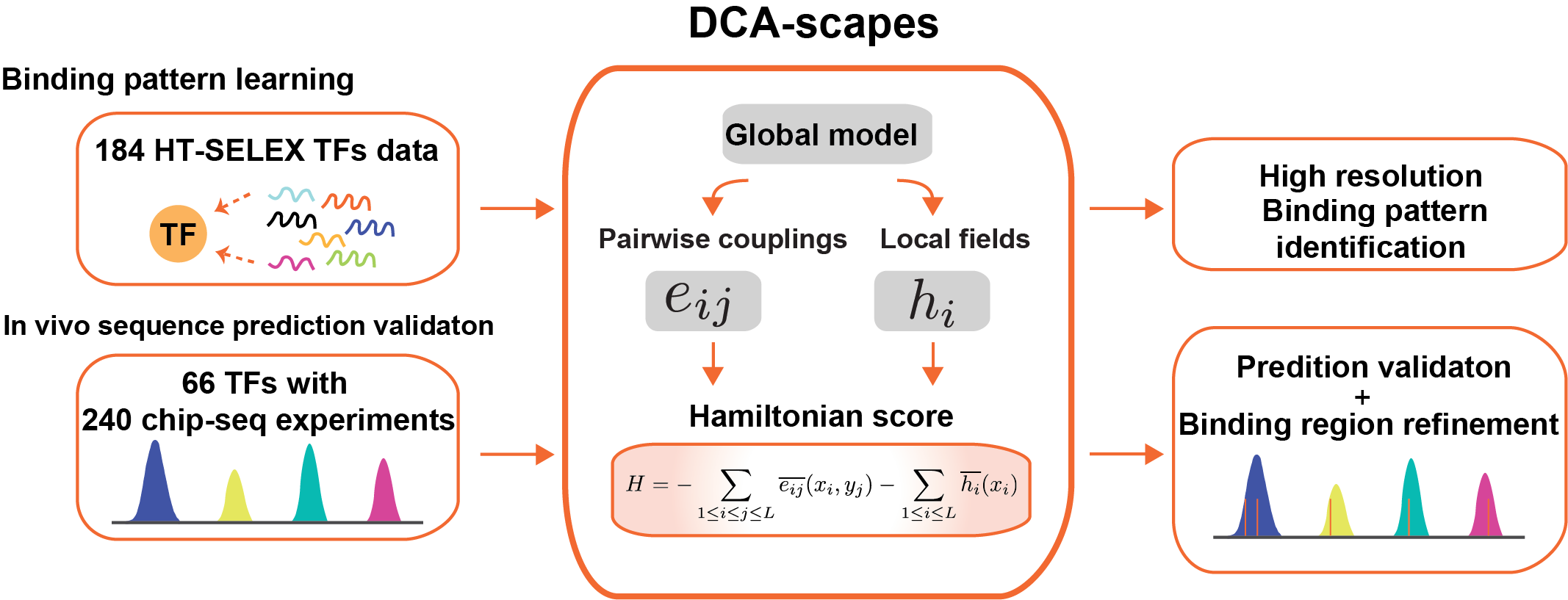
For each TF protein, the coevolutionary model was trained with the TF-bound DNA sequences in round 4 of HT-SELEX experiment, and the initial non-selected sequence pool in round 0 as the background set. With the DNA sequences that selected from HT-SELEX, this model first estimated a joint probability distribution of these sequences with two types of recognition parameters: pairwise couplings eij and local biases hi. For any given DNA sequence, these recognition parameters can be collectively interpreted as the Hamiltonian score (see below sections for details), which quantitatively predicts how likely this DNA sequence binds to the Transcription factor. A random sequence distribution null model that included one million random sequences with similar genomic nucleotide bias was generated to further quantify the probability of interaction. This global comprehensive model was first applied and validated the prediction of exploring the TF-DNA interaction with in vivo binding site of ChIP-seq experiments. Based on the accuracy in Chip-seq data prediction, TFs are separated into two groups: Validated TFs models (validated using ChIP-seq data with high accuracy: average AUC>0.7, N=18) and not-validated TFs models (TFs don’t have ChIP-seq data to compare, N=81 or get low AUC score, N=48).
For the training data (FR) and background data (BG), This model separately infer the joint probability distribution of 20mer DNA sequences with two types of parameters: pairwise couplings eij and local biases (fields) hi :
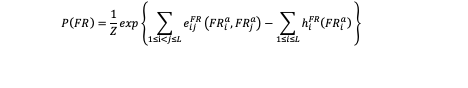
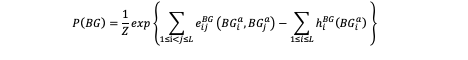
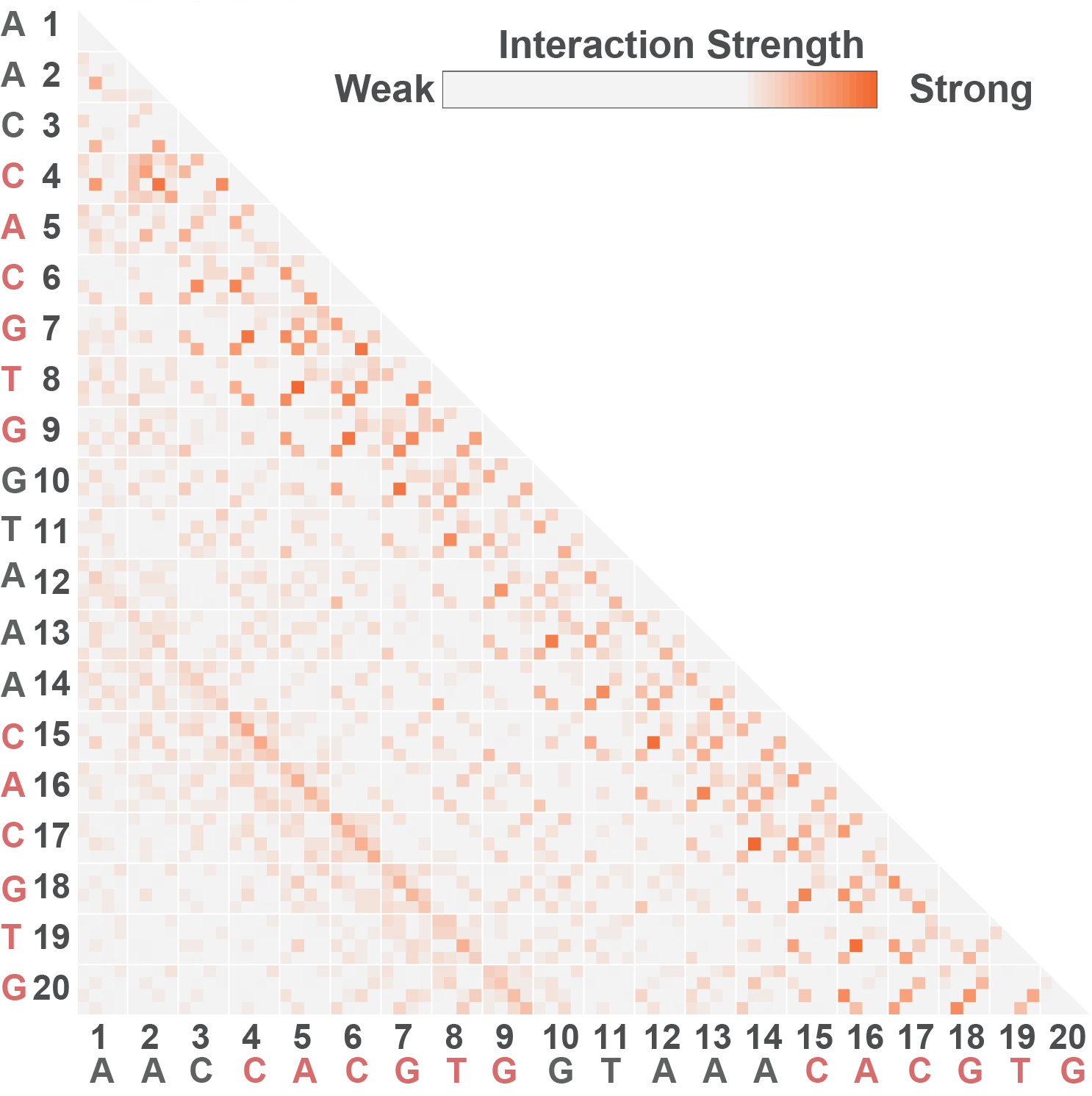
Furthermore, these recognition parameters can elucidate high-resolution maps of specificity to further understand the nucleotide contribution of couplings in TF recognitions.
Protein-binding recognition landscape
For a given DNA sequence x, the Hamiltonian score could quantitatively predict how likely this DNA sequence be a target for the Transcription factors. The more negative Hamiltonian score suggests more favorable TF recognition.
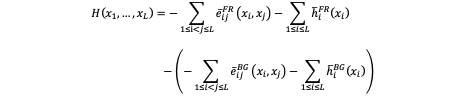
For the given DNA sequence of interested or the entire genome sequence, the Hamiltonian scores were calculated for each 20mer sequences, with a sliding window size of 20, from the first nucleotide to the end of the sequence. For users input DNA sequences, all the Hamiltonian score of every 20mers will be reported with p-value calculated based on the null model. For genome sequences, only the most favorable potential binding sequences are reported.